LCHAD deficiency and extreme prematurity: challenging aspects of nutritional management
In this article we present the case of an extremely preterm female infant who was born at 22+5 weeks’ gestation following an antepartum haemorrhage. A postnatal genetic diagnosis of long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency was made. LCHAD deficiency is a complex metabolic condition, which can be even more challenging to treat when combined with extreme prematurity. Using specialised milk formulas in an extremely preterm infant was a difficult decision; this case report aims to highlight such nutritional and metabolic challenges.
Hiba Ahmed1
Paediatrics Specialty Trainee ST4
hiba.ahmed4@nhs.net
Katherine Pettinger2
Neonatal Registrar
Sally Aitken1
Paediatric Dietitian, Team Leader
Hassan Gaili1
Consultant Neonatologist
1Hull University Teaching Hospitals NHS Trust
2Bradford Teaching Hospital NHS Foundation Trust
Case presentation
An extremely preterm female infant was born at 22+5 weeks’ gestation by spontaneous vaginal delivery following an antepartum haemorrhage and suspected chorioamnionitis. Her birth weight was 510g. The mother had received magnesium sulphate and two doses of antenatal steroids. The baby received surfactant via an endotracheal tube following deferred cord clamping.
Antenatal ultrasound scans had been normal. There was a known family history of LCHAD deficiency; an older sibling has the condition. The mother had a history of HELLP (hypertension/haemolysis, elevated liver enzymes and low platelets) syndrome in a previous pregnancy, as well having lost another previous pregnancy following an amniocentesis for possible LCHAD deficiency. A ‘paediatrics alert’ was written antenatally with a plan for early manage-ment and investigations for the baby.
After admission to the neonatal unit, the baby was started on parenteral nutrition at 90mL/kg/day. On our neonatal unit the standard lipid component is Intralipid, which contains glycerin, soybean oil, and egg yolk phospholipids. However, given the family history, in this case SMOF (soybean oil, medium-chain triglycerides, olive oil, and fish oil) was commenced instead. SMOF has a higher concentration of medium-chain triglycerides compared to other parenteral nutrition lipid products.
After consultation with the metabolic team, trophic feeds were started on day five with donor expressed breast milk (EBM). Maternal EBM was unfortunately not available and there had initially been parental hesitancy in giving consent for donor EBM. Feeds were cautiously introduced at 0.5mL three hourly. This was tolerated well, therefore the feed frequency was changed to two hourly. We were advised by the metabolic team dietitian that EBM should not exceed more than 10% of energy requirements.
Blood tests for LCHAD deficiency were requested on the first day of life. The infant was shown to be homozygous for the common c.1528G>C (p.Glu510Gln) pathogenic variant on the HADHA (hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha) gene, confirming the diagnosis of LCHAD deficiency. These results were available on day nine of life.
Following confirmation of the diagnosis, EBM was stopped and Monogen formula (Nutricia) was started. Monogen is a ‘food for special medical purposes’ that is low in long chain triglycerides and high in medium chain triglycerides. It is not designed specifically for premature infants and does not satisfy the increased nutritional requirements of premature infants for protein and energy.1 It was calculated that the infant would require 180mL/kg/day of Monogen to meet the recommended daily requirement of protein (4g/kg/day). Feeds had initially been given continuously using a pump but were switched to hourly bolus feeds, as we found Monogen formed lumps within the syringe pump.
Like many extremely preterm infants, the patient developed hyperglycaemia, which required an insulin infusion. This dependence on exogenous insulin was unusually persistent; she remained on insulin until day 46 of life. Therefore, a decision was made to reduce her carbohydrate intake while trying to maintain her protein intake. This was initially attempted using a different medium chain triglyceride formula, Lipistart (Vitaflo, Nestle), which has a lower carbohydrate content compared to Monogen. She was commenced on Lipistart on day 59 and was receiving Lipistart and Monogen for one day in the ratio of 1:3 Lipistart:Monogen. However, her blood sugar levels were still rising and the feeds had to be stopped the next day due to an acute deterioration with presumed necrotising enterocolitis (NEC).
Progress
The episode of NEC was managed conservatively and Monogen feeds were gradually reintroduced. Given the previous issue with hyperglycaemia, we gave a total of 150mL/kg/day and added a protein supplement to ensure sufficient protein intake. One sachet of protein supplement provides an additional 0.55g of protein per 100mL of formula.
As with the majority of extremely preterm infants, the child’s neonatal course was complicated. Some of her problems were related to prematurity, some to her LCHAD deficiency and some, a combina-tion of both. Throughout her admission she had respiratory distress syndrome that evolved into chronic lung disease, an episode of suspected NEC, a patent ductus arteriosus, and left ventricular hypertrophy with mild outflow tract obstruction. The echocardiography findings resolved on subsequent scans and were thought to be related to a course of dexamethasone prior to extubation.
Although her serum calcium level was normal, we checked her parathyroid hormone levels as hypoparathyroidism is a known association with LCHAD; in this case, the levels were normal.
At 32 weeks corrected gestational age, the infant was transferred to a regional metabolic centre. There she was found to have developed significant retinopathy of prematurity, which was treated with anti-VEGF therapy. She was discharged home from her local neonatal unit at 40 weeks corrected gestational age weighing 2.34kg, fully bottle feeding on Monogen and receiving home oxygen. Very sadly she died unexpectedly at home shortly afterwards.
Discussion
LCHAD deficiency is a rare form of fatty acid oxidation defect. Mitochondrial oxidation of fatty acids plays an essential role in energymetabolism during periods of fasting. It is an autosomal recessive condition so there may, or may not, be a family history. After birth, when the infant is separated from the placenta, mobilisation of fatty acids is essential to maintain energy supply until enteral feeding is well established. The inability to oxidise fatty acids can result in significant hypoketotic hypoglycaemia during the first days of life. Initial clinical presentation of LCHAD deficiency is variable. Severe hypoglycaemia, cardiomyopathy, encephalopathy, liver dysfunction, myopathy, pigmentary retinopathy, and sudden unexpected death in infancy have been reported.2 Later, patients may develop chronic weakness, pain and recurrent rhabdomyolysis. Acute decompensation may occur with episodes of concurrent illness, decreased oral intake, prolonged fasting episodes, or surgery. As with this family, mothers of affected fetuses can present with acute fatty liver of pregnancy or HELLP syndrome.3
Of course, in normal circumstances preterm infants should be started on breast milk rather than formula. However, the mainstay of treatment for LCHAD deficiency is dietary: a relatively high medium chain triglyceride intake reduces the accumulation of potentially toxic long-chain 3-hydroxy-fatty acids in LCHAD deficiency and that a preparation with a higher ratio of decanoate to octanoate may be most effective.4
The feeding requirements were a maximum of 10% energy from long-chain triglycerides to prevent the accumulation of long-chain triglycerides in stores and accumulation of toxic metabolites, and a minimum of 10% energy from carbohydrates to ensure fat stores are not broken down.
As discussed, Monogen is not a preterm formula. Preterm babies require 110-135kcal/kg/day and 4.0-4.5g/kg/day protein. TABLE 1 demonstrates that high volumes of Monogen would be needed to meet these requirements. Therefore, the baby’s feed was supplemented by the addition of a protein supplement.5 One 1g sachet containing 0.82g hydrolysed whey protein was added to 150mL Monogen to give a total protein content of 2.74g/100mL, resulting in an additional 0.55g protein/100mL. Lipistart has a similar carbohydrate content to standard term infant formula, but Monogen has around 60% more carbohydrate and, therefore, a higher energy content of 74.6kcal/100mL compared with around 69kcal/100mL for Lipistart.6
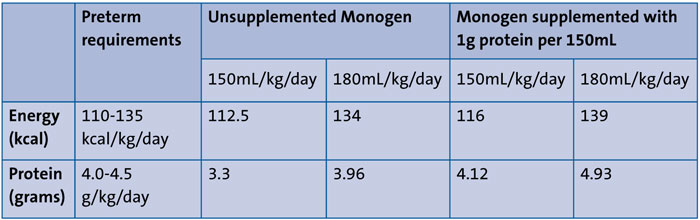
TABLE 1 Nutritional content of Monogen (supplemented and unsupplemented) relative to preterm requirements.
Conclusion
This case shows the challenges of manage-ment of rare metabolic disease such as LCHAD deficiency in an extremely premature infant. Although the family history enabled us to make an antenatal plan including postnatal investigations, the turnover time for the result was still approximately nine days. This made decision making about optimal timing and type of enteral feeds very difficult. Further dilemmas were presented while trying to meet the baby’s protein and energy requirements, while using artificial formulas not specifically designed for preterm babies. Considering the complexity of extreme prematurity and metabolic disorder, multidisciplinary team involvement was a crucial factor in the management of this infant.
Acknowledgement
The authors would like to thank the baby’s family for allowing us to share this case. Thank you to the Sheffield Children’s Metabolic team for their support while writing up this case, in particular Dr Mark Sharrard, for his invaluable expertise and encouragement.
Parental consent
Written consent for publication was provided by the patient’s parent.
Or read this article in our
Tablet/iPad edition
- LCHAD deficiency is a severe and rare form of fatty acid oxidation defect. The primary goals are to prevent fasting and to decrease dietary intake of long-chain fatty acids.
- The coexistence of extreme prematurity and metabolic disorders necessitates multidisciplinary management invol-ving dietitians and neonatal medicine, genetics and metabolic teams.
Also published in Infant: