Facial paralysis in Haddad syndrome: a case report of Ondine-Hirschsprung’s syndrome with a novel clinical feature
Haddad syndrome is a rare congenital disease that results in an autonomic nervous system dysfunction leading to respiratory failure due to loss of vagal input. In this article we report the case of a newborn male with central hypoventilation syndrome and Hirschsprung’s disease who presented with a facial paralysis – a new clinical presentation in Haddad syndrome. As this disease is not common, we describe the process in which the multidisciplinary team approached the patient and discuss the clinical and genetic aspects, aetiology and treatment.
María Camila Martínez Ayala1Intern
mariamaay@unisabana.edu.co
Jaime J. Martínez Arias2
Chief of General Pediatrics
Felipe García Durán3
Medical Student
Alejandro Rueda Rodriguez1
Intern
1Medical School, University of La Sabana, Chía, Colombia
2Department of Pediatrics, Santander Ophthalmology Foundation (FOSCAL), Floridablanca, Colombia
3Medical School, Nueva Granada Military University, Bogotá, Colombia
Background
Haddad syndrome is a rare genetic disorder characterised by the combination of congenital central hypoventilation syndrome (CCHS) and Hirschsprung’s disease. It is seen in approximately 1 in 1,000,000 children and it is associated mostly with genetic mutation on chromosome 4p13; specifically, a heterozygous mutation in the PHOX2B gene, with other genes being associated, such as ASCL1 and RET.1 The PHOX2B gene encodes a transcription factor responsible for the regulation of genes involved in the development of neural crest cells. A mutation in PHOX2B has been detected in 92-100% of cases of CCHS and is associated with Hirschsprung’s disease.2
CCHS was previously known as Ondine’s curse, a name rooted in Nordic mythology – the legend of a water nymph who cursed a man leaving him unable to breathe spontaneously. Described by Robert Mellins in 1970,2,3 CCHS was thought to be an idiopathic condition, but later studies showed its genetic aetiology.1 Concomitant presentation of CCHS with tumours derived from the neural crest and Hirschsprung’s disease was first described by Gabriel Haddad in 1978, hence Haddad syndrome.4
Facial palsy is uncommon but can lead to significant problems in the newborn; the most frequent aetiology is trauma. Other, less common causes are linked to genetic or pathogenic factors. This condition could be classified as unilateral or bilateral, and in some cases, it can occur along with other symptoms, which together make up a syndrome.5
In this article, we report the case of a newborn male who was referred to Clínica FOSCAL in Floridablanca, Santander, Colombia, with a subacute onset of irregular respiratory pattern that required mechanical ventilation, left facial paralysis and seizures that started a few days after delivery. The absence of intrinsic ganglion cells was later confirmed in our institution by anatomopathological studies. We describe the complex multidisciplinary approach that led to the diagnosis of his condition.
The case report
A male neonate of 37 weeks’ gestation was born in a health centre at Valledupar, Colombia, He was delivered via a scheduled caesarean section due to maternal history of antiphospholipid syndrome. He was classified as appropriate for gestational age weighing 2,800g. He had Apgar scores of 6 and 8 at one and five minutes, respectively. His parents were unrelated with a history of three previous miscarriages and one healthy son.
The review of the previous institution’s medical history drew attention to a congenital central facial paralysis on the left side, associated with lagophthalmos and hypotonic extremities. The caesarean section report narrated a procedure without complication and a good technique; there was not any type of trauma to explain the facial palsy. At one hour of age, the medical staff noticed an irregular breathing pattern: slow and shallow breathing when awake but prolonged respiratory pauses when asleep. This progressed to development of central cyanosis, leading to a respiratory failure requiring artificial ventilation with an endotracheal intubation. Three days later, a tracheostomy was performed. The newborn infant was not able to breastfeed due to the mechanical ventilation; continuous orogastric gavage was given. The medical history reported four episodes of generalised tonic-clonic seizures that were treated with phenobarbital, and a suspicion of a thromboembolic event due to the history of the antiphospholipid syndrome in the mother. A computed tomographic scan (CT) was reported as ‘unclear findings of bilateral thrombosis of the transverse sinuses’. On the eighth day, the institution reported that there were signs of infection in the tracheostomy area, a developing fever and toxic appearance; they suspected possible sepsis and started empirical antibiotic therapy with vancomycin plus gentamicin but did not obtain adequate clinical response.
The facial paralysis persisted and the patient was referred to Clínica FOSCAL on his 15th day of life. Here, magnetic resonance imaging (MRI) appeared completely normal, ruling out previous ischaemia or sinus thrombosis suspicion. Other tests for blood group incompatibility and coagulopathy were negative. With regards to the sepsis, a series of blood cultures were performed and were positive for Klebsiella pneumoniae. Piperacillin with tazobactam was initiated. Venous blood gases reported: pH = 7.35; PaO2 = 41.2mmHg; PaCO2 = 57.9mmHg; HCO3 = 31.2mmol/L; base excess = 4.9mmol/L.
On the child’s 20th day of life, he developed abdominal distension. He was still receiving feeds by continuous orogastric gavage but no stool had been passed since day 15. On day 21, a barium enema X-ray was ordered to look for paralytic ileus, volvulus or intestinal atresia (FIGURE 1). The X-ray showed no positive findings but the patient persisted with no intestinal activity. The paediatric surgeon ordered a small intestine biopsy with a local resection of distal intestine (ileum, ileocecal valve, and 5cm of right colon) for pathology studies (TABLE 1). The pathological examination revealed aganglionosis – an absence of ganglion cells. The paediatric surgeon wanted to perform an active search for a viable intestinal section to perform an anastomosis; however, following this procedure, there was still no intestinal activity. The aganglionosis affected the entire digestive tract. The infant remained with orogastric drip-feeding and was ventilator-dependent. Mechanical ventilation weaning attempts were unsuccessful and the irregular breathing pattern persisted.
Three days later the patient developed seizures. The neurology department staff performed video telemetry to investigate brain activity (TABLE 1), which resulted in treatment with levetiracetam to treat the epilepsy.
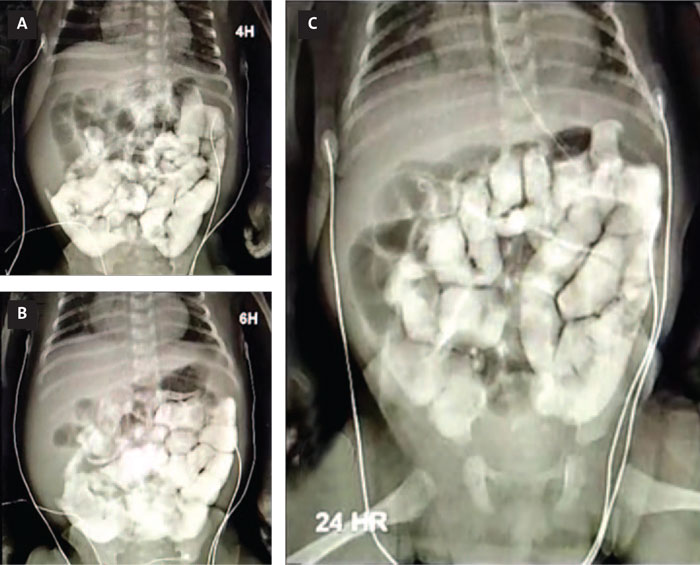
FIGURE 1 The barium enema X-ray. (A) Adequate passage of contrast material through the gastric cavity, permeable pylorus, preserved duodenal mucosa, without extrinsic endoluminal lesions, or malrotations at four hours. (B) At six hours the contrast medium is visualised in the intestinal jejunal and ileal loops, but not seen to arrive at the ileocaecal valve. (C) At 24 hours, persistent contrast medium in the colon and caecum; no abnormal findings.

TABLE 1 Complementary studies.
The pediatrician considered that genetic investigations should be made (eg karyo-typing) but there was no availability for those tests. Given the clinical features of the patient, the medical staff concluded a clinical diagnosis of concomitant CCHS with aganglionosis (confirmed by histopathological studies); the condition known as Haddad syndrome.
After a period of established medical management and several unsuccessful attempts to extubate the patient, an interdisciplinary meeting between the ethics committee and treating physicians proposed that the therapeutic approach should be limited. The parents were consulted at all stages of the clinical process and they agreed that limiting therapeutic efforts would reduce their son’s suffering. The child died one week later at approximately 20 weeks of age.
Discussion
CCHS is a genetic disorder that usually presents in the neonatal period with hypoxaemia, hypoventilation, apnoea, and cyanosis; all predominantly during non-REM sleep, leading to the necessity for assisted ventilation. This can be explained by the pathogenesis of the disorder. PHOX2B expression has been shown to be part of the hypercapnia-sensitive neurons of the retrotrapezoid nucleus; a group of neurons located on the ventral medulla that act as a central chemoreceptor that regulates alveolar ventilation.6
Hirschsprung’s disease is a known gut motility disorder characterised by lack of ganglion cells. A major feature of this disease is that the length of the affected segment is variable. In our case, the clinical presentation was a full-length compromise of the intestinal tract.7
Both CCHS and Hirschsprung’s disease are linked with a group of diverse disorders called neurocristopathies, which result from defective growth, differentiation and migration of neural crest cells. Neurocristopathies can be classified into dysgenetic malformations or neoplastic syndromes,8 and this is reflected in the large spectrum of problems presenting in Haddad syndrome (eg ophthalmic abnormalities, swallowing difficulties, abnormal brainstem auditory-evoked responses, neuroblastoma, facial dysmorphism, hypotonia and fits).9
Current guidelines for the diagnosis of CCHS recommend genetic studies of PHOX2B in patients with Hirschsprung’s disease and hypoventilation;10 however, due to lack of diagnostic resources, molecular diagnosis could not be performed in our case. CCHS was suspected given the presentation of the neonate with hypoventilation and apnoea that were predominant during sleeping periods and associated with central cyanosis with hypercapnia requiring mechanical ventilation. Many of the clinical features present in our patient (eg ophthalmologic abnormalities, hypotonia and generalised tonic-clonic seizures) have been reported in previous reports of the disease, nonetheless central facial palsy has never been described as a clinical manifestation of Haddad syndrome, making this case unique.11
Conclusion
CCHS associated with Hirschsprung’s disease is a rare entity, which should be considered as a differential diagnosis in all newborns or infants who present with respiratory distress and later have difficulty weaning from mechanical ventilation. Our patient presented most of the clinical manifestations of Haddad syndrome plus a novel manifestation; facial paralysis, which warrants further study in the context of this syndrome.
Parental consent
The authors received consent to publish this report from the patient’s parent.
Or read this article in our
Tablet/iPad edition
- Haddad syndrome is a rare condition comprising CCHS associated with Hirschsprung’s disease.
- The patient discussed here presented with most of the clinical features of Haddad syndrome plus a novel manifestation – facial paralysis.
Also published in Infant: