An atypical presentation of Apert syndrome: diagnostic difficulty and the patient journey
Unexpected congenital abnormalities can be associated with diagnostic difficulty and this should be shared with the parents in a supportive way. We present the case of an infant born with congenital abnormalities, the difficulties obtaining a diagnosis and the barriers faced by the parents after discharge from the neonatal unit. Neonatal unit referral processes should be robust enough to provide a safety net in the event of referrals being rejected by secondary or tertiary care; we consider the impact of rejected referrals on parents and how clinicians can support them on the journey to a diagnosis.
Victoria SadlersST5 Paediatrics
Neonatal Unit, Liverpool Women’s Hospital
v.sadlers@doctors.org.uk
Emily Anderson
ST6 Registrar in Clinical Genetics
Liverpool Centre for Genomic Medicine,
Liverpool Women’s Hospital
Chris Parks
Consultant Paediatric Neurosurgeon
and Craniofacial Lead
Department for Craniofacial Surgery,
Alder Hey Children’s Hospital, Liverpool
Unexpected postnatal diagnoses can often cause a great deal of anxiety and it can be difficult for parents to adjust to a new narrative after the birth of an infant requiring unanticipated treatment. Clear communication is paramount in these situations; parents need to know the diagnosis, treatment options and any future management that might be required. This is made more difficult if the diagnosis in question is unknown or uncertain, especially if there were no antenatal concerns.
The practice of neonatal medicine is fraught with uncertainties and many of these uncertainties are managed and communicated effectively and consistently. However, there may be occasions where miscommunication or misinterpretation can result in parents not being fully informed about the clinical situation. Many working diagnoses evolve with time; it is important for clinicians to be open and honest with parents about any diagnostic uncertainty and ultimately this transparency creates trust between parents and clinicians.
We present the case of an infant born with congenital abnormalities and a potential diagnosis of Apert syndrome. There were difficulties obtaining a diagnosis and the parents encountered barriers following discharge from the neonatal unit. The impact of a rejected referral on the parents is considered. We suggest how this situation might have been better handled and how clinicians can support parents on the diagnostic journey.
History
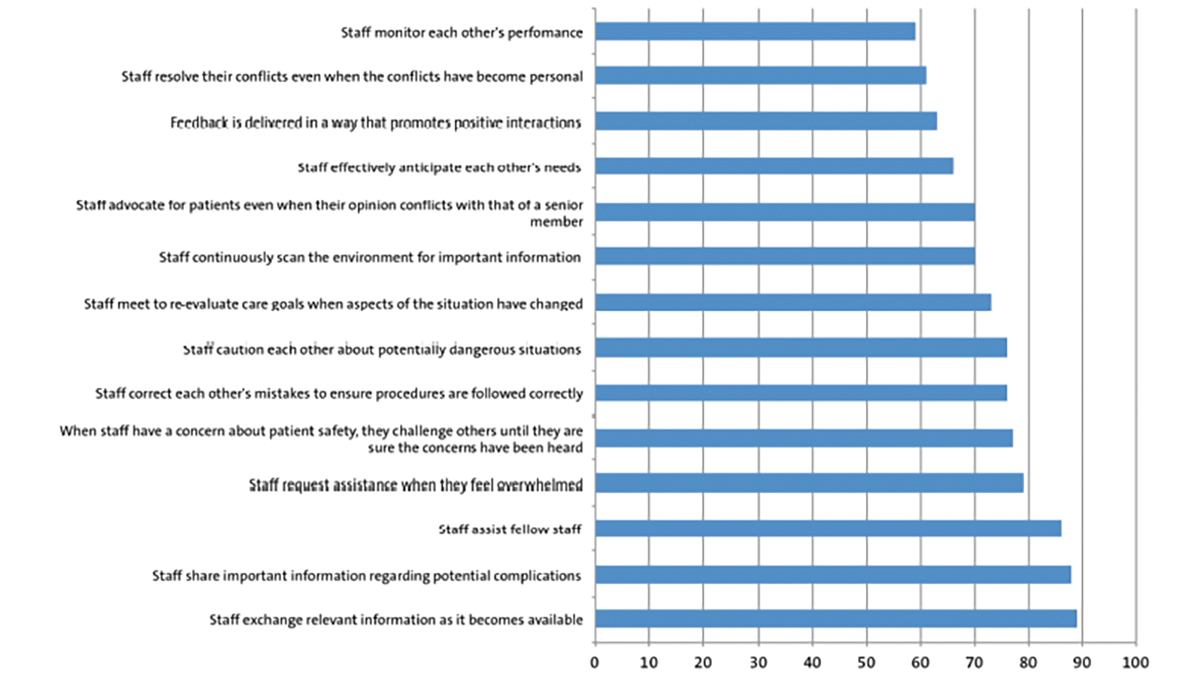
An infant was born at term in unexpectedly poor condition and required full cardiopulmonary resuscitation. Pregnancy history and antenatal scans were unremarkable. During the resuscitation, the patient was noted to have complex bilateral syndactyly of the hands (FIGURE 1) and feet (FIGURE 2). No other abnormalities were noted on examination and the patient had normal cranial morphology.
FIGURE 1 A) Dorsal view of the left hand showing complex syndactyly affecting the second to fourth fingers. B) Palmar view of the left hand showing the abnormal broad thumb with radial deviation and fusion of the second to fourth nails. C) A plain radiograph of the right hand showing the abnormally positioned thumb with radial deviation, fusion of the third and fourth distal phalanges and fusion of the fourth and fifth proximal metacarpals.
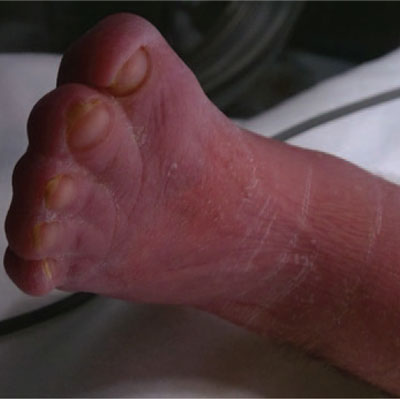
FIGURE 2 Dorsum of the left foot with syndactyly of second to fourth toes with separate nails.
Cardiopulmonary resuscitation was successful and the patient was admitted to the neonatal unit where a nasogastric tube was unable to be passed through either nostril. Therapeutic hypothermia was initiated and the possibility of choanal atresia was discussed. Initially a diagnosis of Apert syndrome was considered, although the absence of any craniosynostosis raised significant doubt.
Members of the neonatal team had cared for infants with Apert syndrome before but not in the absence of craniosynostosis and this prompted discussion among team members as to whether this was the correct diagnosis and how the diagnosis could be made in the absence of the usual criteria. Apert syndrome without craniosynostosis is a rare presentation and only a handful of cases have been reported in the literature.1
The potential diagnosis of Apert syndrome was communicated to the parents, and the diagnostic uncertainty was shared with them at this stage, with a guarded discussion taking place with the parents relating to the diagnosis. The parents were given information leaflets about Apert syndrome and the associated hand abnormalities.
Another complicating factor for this patient was the additional diagnosis of hypoxic-ischaemic encephalopathy and the additional associated uncertainties that the parents had to consider. While still undergoing therapeutic hypothermia, the patient was reviewed by a member of the clinical genetics team who agreed with the likely diagnosis and requested extended genetic analysis of the FGFR2 (fibroblast growth factor receptor-2) gene to try to confirm the diagnosis of Apert syndrome.
The patient was discussed via phone with the regional craniofacial team and a referral letter was sent. Computerised tomography (CT) scanning with 3D reconstruction was undertaken alongside the magnetic resonance imaging (MRI) scan that is required after therapeutic hypothermia. The CT scan revealed patent choanae with no evidence of cranio-synostosis (FIGURE 3). Bilateral dysplasia of the horizontal semi-circular canals and an absent septum pellucidum were also identified. A review of the literature after the event suggests that both of these findings can be associated with Apert syndrome.2,3
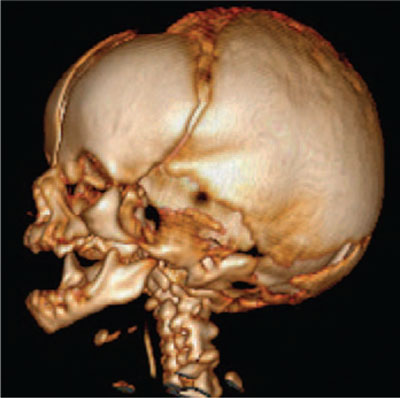
FIGURE 3 3D reconstructed computerised tomography of the skull showing normal cranial morphology and absence of craniosynostosis.
The patient continued to recover on the neonatal unit and was discharged home at three weeks of age after referrals were sent to the tertiary hospital. At the time of discharge the working diagnosis was that of Apert syndrome, and this was communicated with more confidence following multidisciplinary discussions and review by clinical genetics. The likely diagnosis featured on discharge correspondence to the GP.
A few days after discharge from hospital, the parents received an administrative letter from the tertiary hospital advising that the referral to the craniofacial team had been cancelled. The letter did not contain any further information or advice and the parents were understandably upset and anxious upon receiving it. It was not clear why the referral had been cancelled. Fortunately, the patient was under the care of the community neonatal liaison nurse who was able to continue to coordinate care with the other specialty teams required as an outpatient. Upon further investigation after the event, the absence of craniosynostosis meant that the patient did not fit the criteria for an urgent review in the regional craniofacial clinic and the clinic appointment had been cancelled pending diagnostic test results, although, unfortunately this was not communicated to the neonatal team, or the parents at the time.
Shortly after discharge, and after the cancellation letter was received by the parents, the results of the genetic analysis were available and identified a common pathogenic variant in the FGFR2 gene, confirming a diagnosis of Apert syndrome. At this stage the parents were informed and the craniofacial team initiated contact with the parents and arranged for the patient to be seen in the multidisciplinary craniofacial clinic. Due to the complex nature of Apert syndrome and the potential for complications to develop, including postnatal craniosynostosis, the patient requires regular craniofacial follow up. To date, the patient has required regular CT scans but has not required any surgical intervention. The patient will eventually require surgery on the hand and nail abnormalities to improve and preserve function. Physiotherapy and occupational therapy are ongoing, and will be vital to assist with both gross and fine motor development by the provision of specialist equipment.
Discussion
Dealing with diagnostic uncertainty
While it is imperative that clinicians are open and honest with parents about diagnostic uncertainty, it is important to consider the manner in which this uncertainty is communicated. Communicating in a manner that is too vague or ambiguous may cause the parents to lose trust in the clinician, but over-confidence in an unconfirmed diagnosis is misleading for parents and means that they are not fully informed and able to make decisions about their child’s treatment.4
Clinicians all communicate in different ways and parents may sometimes misinterpret this subtle inter-clinician variation. In this situation, when a diagnosis is evolving or uncertain, having a lead consultant who is responsible for communicating to the parents, liaises with other teams and provides follow up after discharge is beneficial. It provides a point of reference and some consistency for the parents during a very uncertain time. Parents can become very upset when a diagnosis changes for their child; it is important for clinicians to specifically communicate that a diagnosis is not confirmed if this is the case.
While a unifying diagnosis is important to the parents and enables some prognostication, there are many patients in whom a diagnosis is never made. The parents should be reassured that this does not preclude those patients from having the correct treatment for their symptoms5 and the same is true for those patients awaiting confirmation of a diagnosis.
‘Rejected’ referrals and the patient journey
As clinicians we should always consider the patient journey and, when referrals are received that are not deemed appropriate, this should be communicated to the parents and the referring clinician in a respectful and sensitive manner. Often parents are copied in to correspondence to primary care and it is important to be mindful of this and not to use language or terminology that would not be used in face-to-face contact. These communications should always be led by clinical members of the team and should explain to both the referrer and the parents the reasons behind the decision not to review the child. The letter should try and avoid using negative language such as ‘rejected’, ‘refused’, and ‘inappropriate’. The response letter should be constructive and inform the referrer which clinician or specialty is the most appropriate to deal with the problem the patient has presented with. The letter should also offer advice regarding the circumstances in which a repeat referral would be warranted.
The referral letter should be seen as the beginning of a dialogue between the referrer and the specialist teams and if another specialist in secondary/tertiary care is more appropriate to review the patient, the letter should be forwarded to that department and the referrer informed.6 The letter should leave the referrer and the parents clear about the next steps. It is easy to see how a standard template letter signed by a non-clinical team member would not be adequate in most situations. Guidance exists to support and educate clinicians about ‘rejecting’ referrals.7,8
Conclusion
Apert syndrome is a rare craniofacial condition. It usually presents with bilateral syndactyly of the hands (typically involving the second to fourth fingers) and feet, alongside multi-suture craniosynostosis, mid face hypoplasia and exorbitism. Apert syndrome without craniosynostosis is a much more unusual presentation and only a handful of cases have been reported in the literature.1
Given that early multidisciplinary input is extremely important and that cranio-synostosis can develop postnatally, we suggest that any infant born with complex bilateral syndactyly affecting the hands and feet should be discussed with the supra-regional craniofacial centre even in the absence of craniofacial abnormalities. In these situations, consideration should be given to early genetic testing, in particular for the two common Apert syndrome variants in FGFR2.
Diagnostic uncertainty should be shared with the parents and there should be a clear follow up plan in place for any infants who do not necessarily follow a standard diagnostic pathway. Follow up should not be dependent on a certain diagnosis being reached and neonatal services should always provide a safety net for patients who may have referrals rejected from tertiary services to ensure they do not get lost to follow up.
Parents are often in a vulnerable situation after their baby has required an unexpected admission to the neonatal unit and are likely to be anxious about their baby’s future, especially when a diagnosis is unknown. It is important that parents are supported along this journey to a diagnosis; being mindful of the language used around parents is very important.
Parental consent
The authors received consent to publish this report from the patient’s parents.
Or read this article in our
Tablet/iPad edition
- Due to the importance of early multidisciplinary input, we suggest all infants who are born with complex syndactyly of the hands and feet are referred for genetic analysis and craniofacial input.
- Clinicians should be mindful of how seemingly inappropriate referrals are communicated to parents and the effects this could have on the patient journey.
Also published in Infant: