A neonate with congenital nephrotic syndrome
A male infant who was born in poor condition exhibited a persistently raised creatinine level over the first 14 days of life but showed no neurological signs of a significant hypoxic insult. A diagnosis of Finnish-type congenital nephrotic syndrome was eventually made by genetic testing. For neonates presenting with unusual or rare underlying pathologies there may be a period of time in which their signs and symptoms are attributed to a more common pathology. This case report illustrates that alternative more unusual diagnoses must be considered and explored when symptoms and results do not follow an expected course.
Jennifer Peterson1
ST6 Neonatal GRID Trainee
Ramiyya Tharumakunarajah1
ST2 Paediatric Trainee
Yasser Masood1
Consultant Neonatologist
yasser.masood@mft.nhs.uk
Susan Kamupira1
Consultant Neonatologist
Nicholas Plant2
Consultant Paediatric Nephrologist
Congenital nephrotic syndromes (CNS) are evident usually within the first three months of life and characterised by proteinuria, hypoalbuminaemia and oedema. Several forms, such as Finnish CNS, follow an autosomal recessive inheritance pattern. Other associated features include prematurity, placento-megaly, fetal distress, low birth weight, failure to thrive and frequent infections.1 If untreated, prognosis is extremely poor with an estimated survival of less than six months of age.2
The case
A male infant was born at 37+1 weeks’ gestation with a birth weight of 3,006g. He was the third child of Caucasian non-consanguineous parents with no family history of congenital abnormalities. Antenatal scans were entirely normal and the pregnancy was considered low risk.
The infant was delivered in poor condition, with Apgar scores of 1, 5 and 5 at one, five and ten minutes, respectively. Venous cord gas analysis showed pH7.3 and a base excess of -6.8. Arterial cord gas was mildly compromised with pH7.09 and a base excess of -8.9. He was intubated at 12 minutes of age due to poor respiratory effort and transferred to the neonatal intensive care unit. His respiratory distress resolved quickly and he was extubated on day 1 and with no further issues.
Initial blood tests showed abnormal renal function and thrombocytopenia, which were attributed to his poor condition at birth. The platelet levels fell to 17x109/L on day 2 of life and then spontaneously recovered to normal by day 7 of life. His initial creatinine level was 123µmol/L, rising to 163µmol/L by day 4. By day 15 it remained elevated at 100µmol/L. Initial urea was 5.6mmol/L, falling to 2.0mmol/L by day 10.
Despite showing no neurological signs of a significant hypoxic insult, the infant’s blood tests remained abnormal with a raised creatinine level. This was monitored over the first two weeks of life in the hope it would improve. However, by day 10 he had increasing peripheral oedema and the creatinine remained >100µmol/L. On further review of his blood tests, the other striking finding was a persistently low serum albumin levels of <10g/L over the first 14 days of life (FIGURE 1).
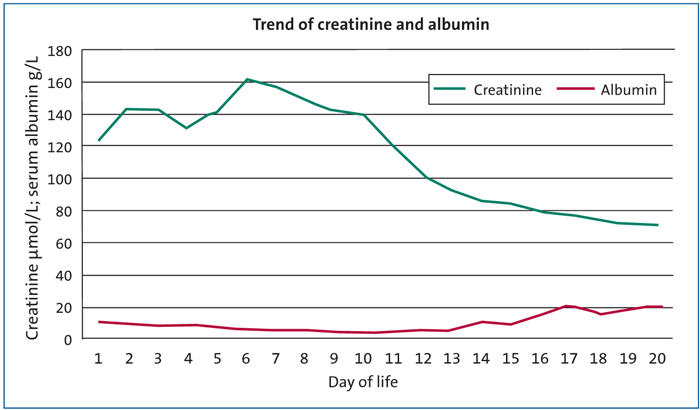
FIGURE 1 The trend of serum creatinine and serum albumin over the first three weeks of life. The diagnosis of Finnish-type CNS was made on day 14.
Further investigation and diagnosis
In view of the child’s normal neurology, low serum albumin and increasing oedema, it was unlikely that his persistently raised creatinine would simply be secon-dary to hypoxic injury. The urinary protein to creatinine ratio was checked and this was found to be raised at 3,243mg/mmol, indicating a CNS. This finding prompted review by the paediatric nephrology team who recommended steroid-resistant nephrotic syndrome genetic panel testing, which revealed that the infant had an NPHS1 gene mutation. The mutation is known to cause Finnish-type CNS.
Discussion
Genetic testing is the diagnostic tool of choice and identification of the NPHS1 gene mutation is the only conclusive method of diagnosing Finnish-type CNS. Having a specific genetic aetiology is important for planning management, discussing prognosis and providing genetic counseling to families.3
Finnish-type CNS is an autosomal recessive condition, presenting in the first year of life. Other characteristic features of patients include the ultrasound appearances of enlarged kidneys with increased cortical echogenicity and loss of corticomedullary border differentiation. Expansion of the glomerular mesangium and dilatation of proximal and distal tubules can be seen on light microscopy, and effacement of podocyte foot processes and absence of slit diaphragm can be identified on electron microscopy, although a renal biopsy is seldom required.4
An antenatal diagnosis of a CNS is suggested by raised amniotic fluid alpha-fetoprotein (AFP) levels due to fetal proteinuria from 15-16 weeks’ gestation. This is often accompanied by an increase in maternal plasma AFP levels. While these are non-specific findings, they may be of use in high-risk cases.5 An important consider-ation is that false positive results have been shown to occur in heterozygous fetuses – one study indicating that out of 21 preg-nancies that had been terminated because of raised concentrations of AFP in amniotic fluid, nine were found to be heterozygotic carriers.6 Heterozygous mutations are associated with a later onset of symptoms as well as a higher likelihood of steroid responsiveness. While follow up data are limited, these individuals are likely to have better outcomes and reduced incidence of progression to renal failure. However, there are heterozygous individuals that will be resistant to steroid therapy, which is a poor prognostic indicator.7
Genetics of CNS
Mutations in the following genes are causative for 85% of patients with CNS:
- NPHS1, encodes nephrin and causes Finnish-type CNS
- NPHS2, encodes podocin (a protein that interacts with nephrin) and is known to cause familial focal segmental glomerulosclerosis
- WT1, encodes a transcription factor that acts as a tumor suppressor and causes Denys-Drash syndrome – a triad of progressive renal disease, male pseudohermaphroditism and Wilms tumour8
- LAMB2, encodes laminin beta 2 (a component of the glomerular basement membrane) and causes Pierson syndrome, a condition characterised by CNS and bilateral microcoria (small pupils)9
The NPHS1 and NPHS2 mutations are responsible for 95% of all cases of CNS caused by gene mutations.10
The patient in the case presented here was diagnosed with Finnish-type CNS. The responsible NPHS gene is found on chromosome 19q13.1 and encodes nephrin, a cell adhesion molecule. Nephrin is produced by the glomerular podocytes and found at the slit diaphragm. Its function is essential to glomerular filtration, acting as a barrier and excluding serum albumin and other plasma macromolecules in the formation of urine.11 The two most common mutations of NPHS1 result in short, non-functional nephrin. The first mutation, known as Finn major, is responsible for 78% of cases, and the second, Finn minor, is responsible for 16% of cases.4
Infection-related CNS
Congenital infections can be the underlying mechanism for CNS in some infants, however congenital infections account for a smaller number of cases than gene-related CNS:
- congenital syphilis can result in a membranous nephropathy, which responds well to treatment with penicillin12
- congenital toxoplasmosis can cause a nephrotic syndrome associated with ocular and neurological symptoms. In most of these cases, treating the toxo-plasmosis will resolve the proteinuria, however some patients may require steroid therapy13
- other cases of congenital nephrotic syndrome have been attributed to viral infections, such as cytomegalovirus, rubeola virus and HIV.14
Management
As Finnish-type CNS is always resistant to glucocorticoids and immunosuppressive treatment, long-term management poses many challenges.15 Management can be problematic and with huge implications for the family. Generally, patients are managed with regular albumin infusions, often many times each week, until they have unilateral or bilateral nephrectomies. With one kidney removed, their albumin requirements are usually reduced, making management slightly easier, and the second kidney is removed a few months later. After bilateral nephrectomies, renal replacement therapy is required until transplantation is possible. Many infants will also need treatment with angiotensin converting enzyme and prostaglandin inhibitors, as well as diuretics, thyroxine and a degree of anticoagulation. Most infants will spend the majority of their first year of life in hospital in order to receive this care.
Regular albumin transfusions require long-term central venous access with the inherent dangers, predominantly infection and thrombosis, that this access entails. Dialysis poses its own difficulties with access and infection risks and is usually only attempted once the infant is at least 5kg. Renal transplantation is possible when the child reaches 10kg and a suitable donor is available.16
There are huge practical, financial and psychological implications for the family with all of the management options. In addition, the prognosis in CNS with and without intervention is poor. Families, including siblings, benefit from psychology support.
Outcome
The infant in this case study is now 18 months old. He needed regular albumin infusions up until two months ago. His estimated glomerular filtration rate is now markedly reduced (25mL/min/1.73m2). The plan is for him to undergo bilateral nephrectomies followed by peritoneal dialysis.
Prognosis
Overall, this diagnosis carries a poor prognosis. A recent study by Berody et al17 followed a cohort of 55 patients with CNS who had a variety of underlying genetic causes (the majority had NPHS1). Diagnosis of CNS was made at a median age of 14 days of life. Thirty-nine patients reached end-stage kidney disease at a median age of 11 months. The overall renal survival was 64% and 45% at one and two years of age, respectively. Patients must then be suitable for, match and undergo renal transplantation, with the plethora of short- and long-term risks this carries. The study did find that renal and patient survivals were longer in patients with NPHS1 mutations than for patients with other underlying genetic mutations.
Conclusion
It is extremely important to further investigate symptoms that do not fit with an initial diagnosis. In this case, the initial high creatinine could have been due to hypoxic injury at birth but it did not follow the usual course and improve over first few days. This and the very low serum albumin led to further investigation, ultimately leading to the diagnosis of Finnish-type CNS.
Parental consent
The authors received written consent to publish this report from the patient’s parents.
Or read this article in our
Tablet/iPad edition
- It is important to further investigate any symptoms that do not fit with an initial diagnosis.
- The genetic basis for congenital nephrotic syndrome (CNS) is becoming better defined and testing is more readily available.
- The NPHS1 gene encodes nephrin and is implicated in autosomal recessive Finnish-type CNS.
- CNS can also be caused by genetic defects in NPHS2, LAMB2 and WT1 genes.