A rare and unusual presentation of aplasia cutis congenita
Aplasia cutis congenita (ACC) is a heterogenous group of disorders characterised by the absence of a portion of skin in a localised or widespread area at birth. ACC manifests as a solitary defect on the scalp in the majority of cases, however, extensive lesions on the trunk and limbs are reported in the literature as rare presentations. Most cases are sporadic but ACC has also been associated with twin pregnancies, in which one twin died during early pregnancy. In this case study, we report a rare case of extensive cicatricial ACC on the trunk and lower limbs of a male infant.
Marika LasokovaMD, MRCPCH, MSc Science
Paediatric Specialty Registrar
marika.lasokova@nhs.net
Ourania Kaltsogianni
MD, MRCPCH, MSc Advanced Paediatrics
Neonatal GRID trainee
Jogesh Kapadia
MBBS, DCH, DNB(Paeds), MRCPCH, FHEA
Dip Clin Risk & Claims Mngmt, Masters in Med Ed
Consultant in Neonatal Medicine
Luton and Dunstable University Hospital NHS Trust
Background
ACC is a rare condition with an incidence of 1-3 per 10,000 live births.1 Most cases are sporadic but familial cases occur in an autosomal dominant or recessive inheritance pattern. Most lesions occur on the scalp lateral to the midline, but they may also occur on the face, trunk, or limbs. Extensive lesions on the trunk and limbs have been described in association with twin pregnancies, in which one twin died during early pregnancy and remnants of the twin (fetus papyraceus) were found in the placental membrane.2-4
The case
A term male infant was born at 39+3 weeks’ gestation by planned caesarean section for breech presentation. He was the surviving monochorionic diamniotic (MCDA) twin; the other twin’s demise occurred at 16 weeks’ gestation. Other than this, the pregnancy was uncomplicated and there was no evidence of fetus papyraceus at delivery. The child was born in good condition and did not require breathing support at any point during his stay. There were no cardiovascular concerns.
At the time of birth, an extensive, well-demarcated, symmetrical, stellate-shaped lesion was noted in the sacro-lumbar area extending into the lower limbs (FIGURE 1). The lesion appeared silky, transparent and thin with an absence of skin layers. The anus was placed posteriorly. There were no other obvious associated anomalies. Clinically, spina bifida could not be excluded with certainty at this stage. X-rays of the chest and abdomen were normal.
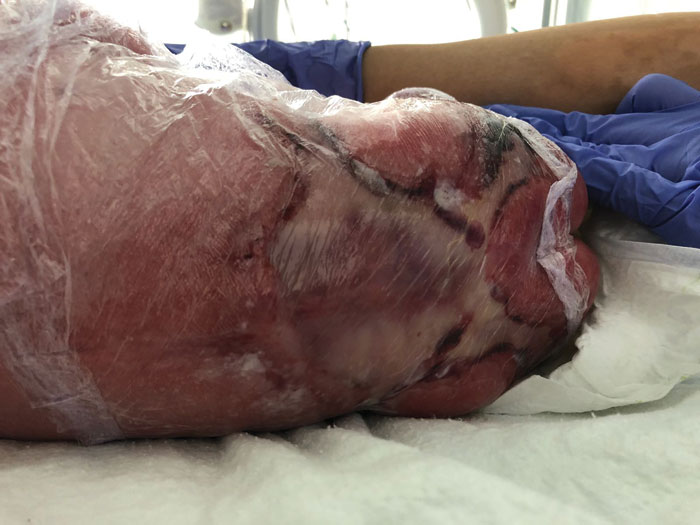
FIGURE 1 The extensive ACC lesion over the lumbosacral area at birth (wrapped in cling film).
A blood culture was negative and so were inflammatory markers. Histology was not performed.The skin over the affected area was covered by cling film and the infant was transferred to a tertiary centre for a multidisciplinary team review. Myelomeningocele was excluded. The infant was reviewed by the surgical team due to the posterior anus. Regular bowel passage provided further assurance.
The lesion was not thought to be part of a genetic syndrome and conservative treatment was recommended, initially cream and sterile dressings followed by scar massage and silicon spray with advice on compression garments and silicon pads. The lesion gradually healed without any complications.
Although there was no evidence of fetus papyraceus at delivery, placental histology revealed infarcts involving 15% of the placental disc.
Discussion
Although small, solitary scalp ACC lesions are frequently reported, such an extensive lesion on the trunk and lower extremities, as seen in this patient, is relatively rare. ACC can be located anywhere on the body and its clinical appearance and location can alert the clinician to other potential abnormalities and associations. In 1986 Frieden classified ACC into nine groups according to localisation and associated malformations (TABLE 1).4,5
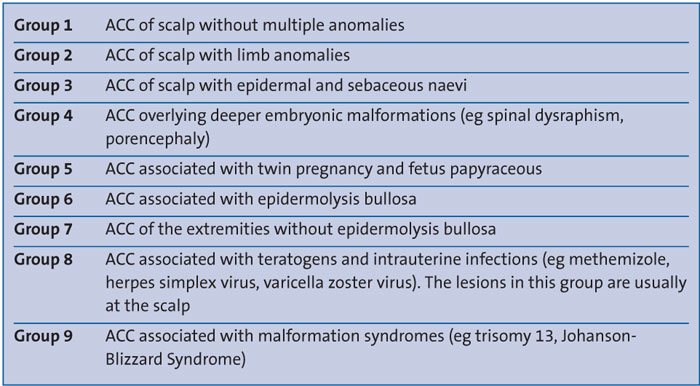
TABLE 1 The classification of ACC based on aetiology and associated anomalies according to Frieden and described by Gupta et al.4,5
Frieden described Group 5 lesions as truncal ACC with symmetrical absence of skin in a stellate or butterfly pattern with or without involvement of the proximal limbs, and associated with fetus papyraceus. It is the most common congenital cicatricial (formation of a dense fibrous tissue resulting in a scar) alopecia and is a congenital focal absence of epidermis, with or without evidence of other layers of the skin. The patient presented here had lesions conforming to this description; however, there was no evidence of fetus papyraceus.The aetiology of ACC is still unclear and it is likely that there are several potential causes for its development including intrauterine infection, medications during pregnancy (eg methimazole, misoprostol, valproate, cocaine, marijuana), fetus papyraceus, feto-fetal transfusion, vascular coagulation defects, amniotic membrane adherence, abnormal elastic fibre, biomechanical forces and trauma. It can also be associated with rare genetic syndromes and has been linked to peptidase D haploinsufficiency and a deletion in chromosome 19.6,7 Non-spontaneous ACC can be induced by selective fetal reduction.3
Many of the sporadic cases of ACC have been associated with placental infarcts or twin pregnancies, in which one twin died during the late first or early second trimester. In these cases, the pathogenesis may be related to vascular disruption inducing abnormal dermoepidermal development and cutaneous defects through ischaemic and thrombotic events. The intrauterine death of one of the fetuses could cause the release of thrombosis-promoting substances that can cause placental infarction, disseminated intravascular coagulation and cutaneous lesions.4 Lesions that form early in gestation may heal before delivery and appear as an atrophic, membranous, parchment-like or fibrotic alopecic scar. Less mature defects may present as an ulceration of variable depth. When just the epidermis and the upper dermis are involved, minimal alopecic scarring may result. Deeper defects may extend through the dermis, subcutaneous tissue, periosteum, skull, or dura.8
Conclusion
This rare disorder can have varied manifestations and requires a thorough investigation to achieve an accurate diagnosis and classification. It is important to exclude other associated malformations and any underlying genetic conditions. Treatment is usually conservative; skin or bone grafts may be used for the reconstruction of major defects. The majority of lesions resolve fully within the first few months of life, leaving behind little evidence.
Parental consent
The authors received written consent to publish this report from the patient’s parents.
Or read this article in our
Tablet/iPad edition
- Extensive ACC lesions on the trunk and limbs are rare.
- ACC can be associated with placental infarcts or the in utero death of a twin fetus, as in the case presented here.
- Obstetric history should review maternal medications/infections during pregnancy, determine an initial multiple pregnancy with death of a co-twin and investigate any placental anomalies.
- Despite their large size, truncal and limb ACC lesions usually resolve within the first few months of life.
Also published in Infant: