A case of Goldston syndrome in a newborn with a CEP290 ciliopathy
This article describes the case of a live male infant delivered at 34+5 weeks’ gestation in view of antenatally diagnosed abnormalities, including bilateral ventriculomegaly with hydrocephalus and bilateral polycystic kidneys. Postnatally, he was confirmed to have a Dandy-Walker malformation and autosomal recessive polycystic kidneys, the combination of which is suggestive of Goldston syndrome. Trio-exome genetic testing was performed on this infant, revealing a homozygous pathogenic CEP290 variant. We therefore present the first case of Goldston syndrome associated with a CEP290 variant.
Arameh Aghababaie1a.aghababaie@nhs.net
Bijaya Chowdhury1
Nina Tanna1
Ariane Waran2
Lewis Pang3
Aruj Qayum1
1Barts Health NHS Trust, London
2Great Ormond Street Hospital for Children NHS Foundation Trust, London
3Royal Devon University Healthcare NHS Foundation Trust, Exeter
Aghababaie A., Chowdhury B., Tanna N., Waran A., Pang L., Qayum A. A case of Goldston syndrome in a newborn with a CEP290 ciliopathy. Infant 2024; 20(3): 73-77.
Background
Ciliopathies are genetic disorders arising from defective ciliary function or structure, causing a range of developmental abnormalities, most commonly affecting the retina, kidneys and central nervous system.1 Several well-recognised ciliopathy syndromes are described in the literature, namely:2
- Joubert syndrome (JS)
- Bardet-Biedl syndrome (BBS)
- Meckel-Gruber syndrome (MGS).
These are all linked to variants of the CEP290 gene. Although their presentations significantly overlap, each has a distinct set of symptoms.2
Goldston syndrome (GS), first described by Goldston in 1963, is very rare and considered a less severe variant of MGS.3,4 It is characterised by the combination of Dandy-Walker malformation (DWM) with polycystic kidneys, with or without hepatic fibrosis and oligohydramnios.4 Very few cases have been reported worldwide and diagnosis is made clinically, with no known genetic associations.4
We present a case of GS in an infant presenting with DWM and bilateral renal cysts with a confirmed homozygous pathogenic CEP290 variant, making this the first case of GS with a genetic association.
Case presentation
A live male infant weighing 2,585g (92.5th centile) was born at 34+5 weeks to a 28-year-old gravida 1 para 0 female via elective caesarean section in view of antenatally diagnosed abnormalities, including bilateral ventriculomegaly with hydrocephalus and bilateral polycystic kidneys. The mother was partially deaf and the parents were consanguineous.
Antenatal scans showed severe bilateral ventriculomegaly with a posterior fossa defect leading to a communicating hydrocephalus between the third and fourth ventricles, a collapsed stomach and bladder, and bilateral echogenic polycystic kidneys occupying most of the abdominal cavity, with oligohydramnios (likely secondary to impaired renal function). The fetal echocardiogram and Doppler ultrasound were normal.
In light of this, a provisional palliative care plan (PCP) was agreed between the parents, the obstetric team and the fetal medicine team, antenatally. A decision was then made to deliver the baby early because of the increasing growth velocity of the fetal head. To assist delivery of the head, cerebrospinal fluid was drained via the fontanelles. He was born in poor condition with Apgar scores of 2, 4 and 7 at one, five and 10 minutes of age, respectively. The presence of a neonatal team was not requested at the delivery as the infant was not expected to show signs of life once delivered, or at most, survive beyond a few hours. He was brought to his mother to ensure maximal time together, as per parental wishes. However, the infant continued to show signs of life at four hours of age, therefore neonatal input was sought.
On examination, his respiratory rate was 67bpm, with mild intercostal recessions. The anterior and posterior fontanelles were large, tense, and full. His head circumference was 39.5cm (>99th centile) and there was bilateral leukocoria. He was alert but not particularly active and he had a moderate suck. His skin was pale and his temperature was 36.1°C. The remainder of the examination was normal.
He was later admitted to the (level 2) neonatal unit for further investigations and management.
Investigations
The following minimally invasive investi-gations were performed with the intent of understanding the infant’s current medical state, diagnosis and prognosis, while respecting the PCP agreed with the parents antenatally.
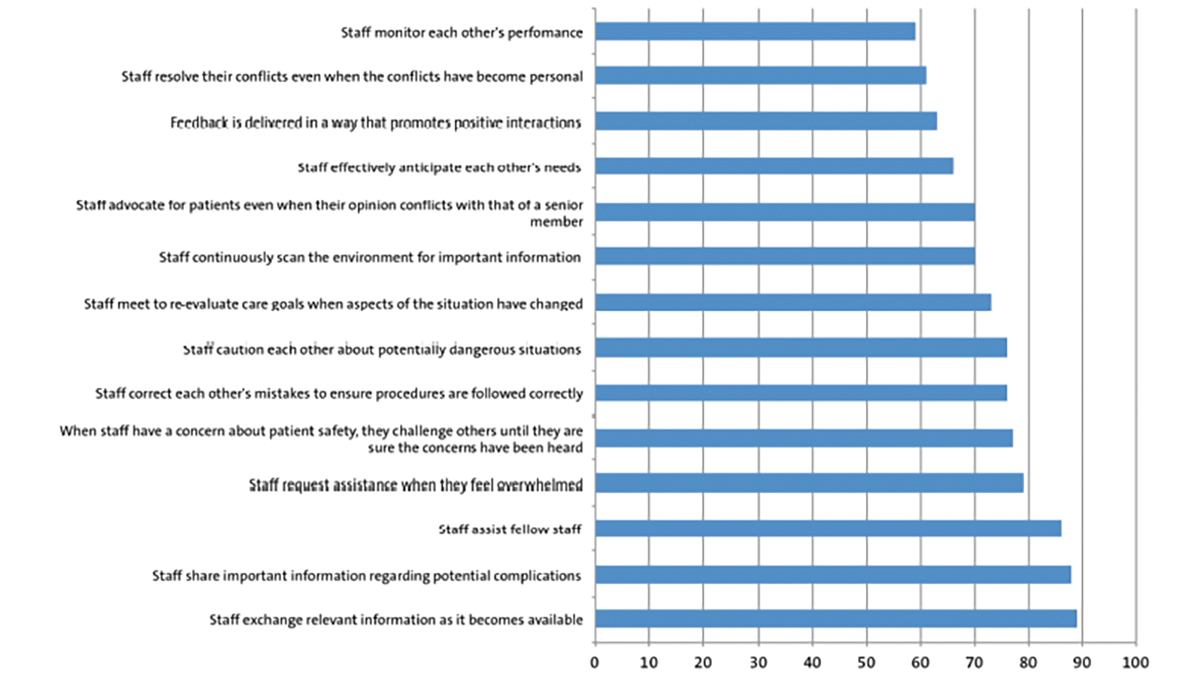
The following blood investigations were performed: capillary blood gas test, urea and electrolytes, bone profile and parathyroid hormone level. The results are presented in Table 1.
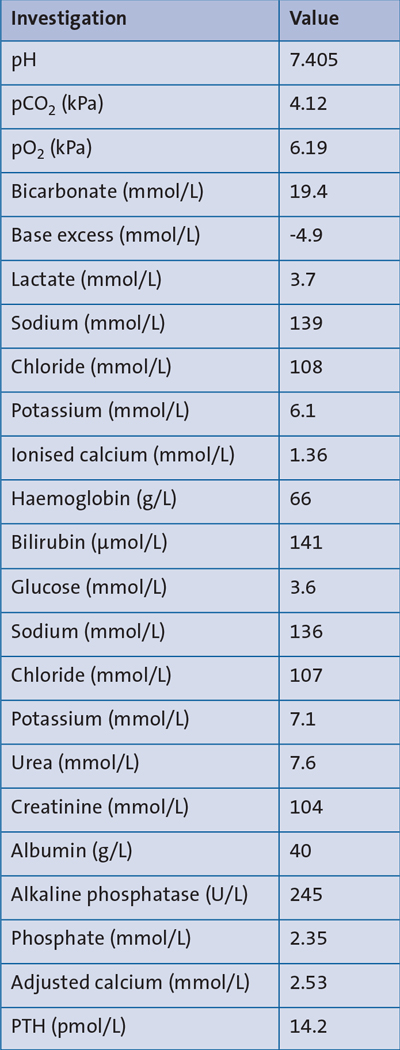
TABLE 1 Results of baseline gas and blood investigations. These results show a high-normal calcium, raised ionised calcium, high-normal alkaline phosphatase, raised parathyroid hormone level (PTH), raised potassium, normal pH, low bicarbonate, and low haemoglobin. This is in keeping a chronic renal failure picture, due to renal impairment of bicarbonate absorption, potassium secretion, erythropoietin production, vitamin D activation and phosphate excretion, leading to secondary hyperparathyroidism.
An abdominal X-ray showed an appropriately sited nasogastric tube (NGT) and a normal intestinal gas pattern throughout (FIGURE 1).
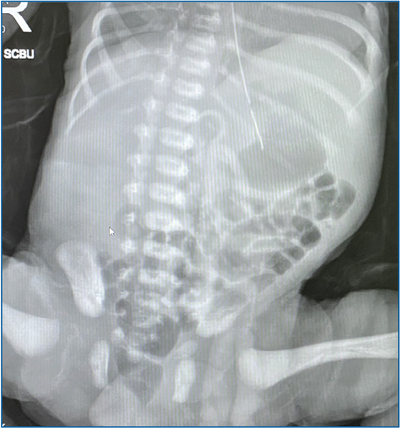
FIGURE 1 An abdominal X-ray taken following NGT insertion. The X-ray shows an appropriately sited NGT in the stomach with a normal intestinal gas pattern throughout.
A cranial ultrasound showed an absent corpus callosum, a small cerebellar vermis and a communicating hydrocephalus between the third and fourth ventricles, with a posterior fossa cyst, suggestive of DWM (FIGURE 2).
FIGURE 2 Cranial ultrasound: a mid-coronal image showing a large communicating hydrocephalus between the third and fourth ventricles, with compressed lateral ventricles. There is also an absent corpus callosum and in the sagittal images (not shown), a small cerebellar vermis is noted.
An abdominal ultrasound showed bilateral large kidneys with innumerable parenchymal cysts, more suggestive of autosomal recessive polycystic kidney disease (ARPKD) than cystic dysplasia (FIGURE 3). The bladder was empty but normal in size.
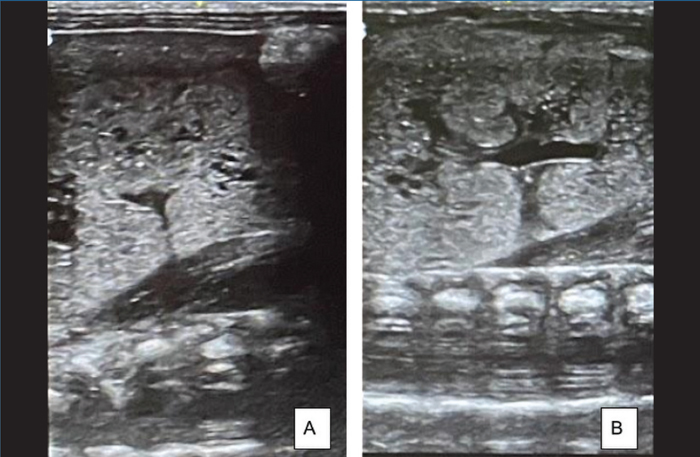
FIGURE 3 Abdominal ultrasound. (A) is an ultrasound scan of the left kidney and (B) of the right kidney. The left kidney measures 4.3cm and the right kidney measures 4.2cm. Both kidneys demonstrate multiple tiny parenchymal cysts suggestive of autosomal recessive polycystic kidney disease. There are no renal stones or hydronephrosis or masses. The remainder of the scan (not shown), revealed a normal liver, gallbladder, spleen and an empty normal-sized bladder.
Differential diagnosis
There was no postnatal evidence of the antenatally-diagnosed collapsed stomach bubble and collapsed bladder. However, the postnatal investigations confirmed the antenatal findings of bilateral ARPKD and DWM.
These findings were initially treated as two separate entities in the child; however, upon a literature review, the rare syndrome known as GS appeared to marry the two entities. Therefore, we referred the infant’s blood samples to the genetics lab for testing, with parental consent. Given the clinical context and background of consanguinity (raising suspicions of a recessive genetic disorder), the genetics team arranged for trio-exome testing using DNA extracted from the blood samples of the infant and the parents (sequencing the protein-coding regions of the genome).
Treatment
The infant received formula feeds via an NGT and received oxygen via a nasal cannula due to gradual intermittent desaturations (as low as 80%).
In light of the investigation results, he was discussed with our neurosurgical centre and tertiary renal and palliative care teams. It was jointly decided that it was in his best interests to continue active treatment at this stage and transfer him to a tertiary unit.
Outcome and follow-up
Following transfer, he had multiple significant apnoea events prompting a series of further investigations, leading to an emergency ventriculoperitoneal (VP) shunt insertion. He had a shunt revision the following week due to continuing significant apnoeas and he developed a thrombus from a femoral central line. He remained in hospital for a further month following the operations due to difficulty in weaning respiratory support and establishing feeds.
The results from the trio-exome testing identified a homozygous pathogenic CEP290 frameshift variant, NM_025114.4:c.3157del p.(Val1053Phefs*7). Both parents are heterozygous carriers of the variant. The variant is classified as pathogenic according to the American College of Medical Genetics and Association for Clinical Genomic Science guidelines.5,6
At one month into the hospital admission, a multidisciplinary team (MDT) meeting was held between the neonatologists, neurosurgeons, neurologists, the renal team, palliative care team and the parents, to discuss the infant’s prognosis and ongoing care.
The neurosurgeons felt his apnoeas were secondary to his posterior fossa pathologies and that his prognosis was very poor. Despite the infant producing urine and maintaining his blood pressure and safe electrolyte levels without medications, the nephrologists felt his co-morbidities made him an unsuitable candidate for renal transplantation. Therefore, a joint decision was made in the infant’s best interests to redirect back to a PCP. He was discharged home on bilevel positive air pressure therapy (BiPAP) and nasojejunal feeding, with a plan to come off BiPAP at home, with support of anticipatory medications.
Discussion
Ciliopathies are caused by variants in ciliary genes, including the CEP290 gene (centrosomal protein 290).3 Pathogenic CEP290 variants are generally associated with eye, kidney and liver disease, along-side features specific to various syndromes, such as Senior-Loken syndrome, JS, BBS, and MGS.2,3 MGS is a rare and lethal disorder inherited in an autosomal recessive manner and associated with:2,7,8
- occipital encephalocele or other posterior fossa abnormalities
- post-axial polydactyly
- hepatic fibrosis
- bilateral multicystic kidney dysplasia.
Goldston syndrome (GS)
MGS has a milder variant known as GS, which is the likely diagnosis in this case.9 No specific genetic variants have yet been identified for GS, until now.
GS is a rare disorder encompassing ARPKD and DWM, which were key features in this case.10-13 Further associations include oligohydramnios (as in this case), polyhydramnios, hypoplastic lungs, gut duplication and hepatic fibrosis.14,15 In this case, there was no polydactyly or occipital encephalocele, differentiating it from MGS.
GS was first described by Goldston et al in which three siblings, two males and one female, were reported to have the condition.4 Since then, a further five fetal cases, two neonatal, six paediatric, and one adult case of GS have been reported.4,10-14,16-19 Therefore, this is the 18th case reported in the literature and the third case in a newborn.
The only case identified in an adult was reported in 2006.11 The case describes a 32-year-old otherwise well female with acute-onset headache, vomiting and truncal ataxia with no clear precipitants and otherwise normal neurological examination. Magnetic resonance imaging (MRI) showed DWM with bilateral subdural frontal collections extending temporally, and an abdominal ultrasound scan showed bilateral polycystic kidneys, with evidence of chronic renal failure in the biochemistry. No hepato-pancreatic abnormalities were found. Karyotyping was normal but no other genetic testing was described.
Of the neonatal cases published, Gulcan et al reported a case of polycystic renal disease, DWM and congenital hepatic fibrosis in a male preterm infant suspected to be GS.16 Agrwal et al also report another case of a 22-day-old male infant with DWM and polycystic kidneys presenting with symptoms of cholestasis.10 The infants improved with supportive management, but it is not stated whether they survived.
The four reported fetal cases share features suggestive of DWM, polycystic renal disease and oligohydramnios.12,13,17,18 Gloeb et al report additional findings of cystic hepatic lesions on post-mortem in their case, suggestive of congenital hepatic fibrosis, which may be the cause of the fetal ascites in Rathod et al’s case.17,18 Antenatal diagnosis was made following ultrasound scans on weeks 17, 22, 24 and 27 of gestation, in 12, 13, 17, 18 respectively.
The other cases reviewed have not explicitly been diagnosed as GS, however, they do outline its hallmark features of DWM with cystic renal disease and occasional cystic hepatic involvement.14,19
The CEP290 gene
Interestingly, the CEP290 gene is important for ciliary function, which enables our vision, hearing and smell.20 This may explain why leukocoria was an additional feature identified in this case of GS. This has not been described in other cases of GS and not thought to be a typical feature. However, pathogenic CEP290 variants are linked to cases of blindness and other retinal pathologies, so it is plausible that this variant may cause retinal pathology.2,3,20 Additionally, the trio-exome results showed a homozygous pathogenic CEP290 variant and the parents were consanguineous. The parents were also both identified as heterozygous carriers, perhaps explaining why the mother was partially deaf.
While there are no definitive diagnostic investigations outlined for GS, once DWM is identified a detailed review of extra-cranial malformations should be performed, particularly with regards to the cardiac, skeletal, gastrointestinal and genitourinary systems.11 Avcu et al suggest correlating abdominal distension found antenatally on ultrasound, with a fetal MRI.12
This is the first case in which a gene has been identified. As GS is a variant of MGS, and MGS is known for being the most severe form of the ciliopathies and has an autosomal recessive inheritance pattern, it is reasonable to suggest that GS too is an autosomal recessive condition with links to ciliary gene variants, including a CEP290 variant. There are links to consanguinity, such as in this case and other cases, and there may be links to abnormal sex chromosomes in parents.14,17 There are several reports of siblings sharing features of GS, supporting an autosomal recessive inheritance pattern.4,14
This is the first case of GS where trio-exome testing has been reported. The advent of next-generation sequencing technologies has meant that it is increasingly common to sequence the whole exome or genome of a patient, when referred for genetic testing. Previously, it was standard practice to sequence a specific gene or selection of genes related to the patient’s phenotype. However, sequencing the exome or genome of a patient and their unaffected parents allows for gene agnostic inheritance-based variant filtering in which the patient DNA is compared against a reference genome to identify variants. These variants are then further filtered based on their mode of inheritance from the parents, regardless of which genes the variants lie in. A major advantage of this method is that it will detect variants in genes that a clinician may not have previously considered. This method was invaluable for this patient as a variant in CEP290 was not predicted prior to the genetic testing. This variant is not detectable by microarray or karyotyping. Therefore, in suspected cases of GS, CEP290 sequencing, whole exome sequencing or whole genome sequencing should be considered, as guided by the genetics team. This can also be useful with regards to planning future pregnancies.
Management
With regards to management, supportive management of individual aspects of the child’s condition generally produces good results.10 In this case, we opted to treat each component of the infant’s condition, in conjunction with our tertiary neurosurgical, renal and palliative care teams, before transfer to the tertiary unit for further intensive review and management.
Despite intensive review and management, the prognosis for GS is poor and the life expectancy is generally hours to months. Of the available literature, only one case is confirmed to have survived to adulthood.11 Having additional hepatic or pancreatic involvement in GS is not known to affect the prognosis and little is known as to which features, if any, determine survival into the neonatal period.10 In this case of GS, a PCP was in place due to recurrent central apnoeas, despite insertion and revision of his VP shunt. Nevertheless, at five months of age he remains alive, likely due to the intact brain stem and respiratory centre, which can prolong life despite multiorgan failure.
Conclusion
This is the first case of GS with a patho-genic homozygous variant identified, specifically in the CEP290 gene. Although ciliary gene variants are commonly associated with its more severe variant, MGS, the clinical features in this case are more in keeping with GS. In future suspected cases, the following should be considered:
- antenatal and postnatal genetic testing
- fetal MRI
- early antenatal and postnatal MDT input, including the neonatal, neuro-surgical, renal, genetics, and palliative care teams
- access to psychological support for the parents.
Learning points
- GS is a rare syndrome characterised by DWM and polycystic kidneys, with occasional reports of oligohydramnios and hepatic fibrosis. This is the 18th case reported in the literature and the third neonatal case reported.
- GS is inherited in an autosomal recessive manner and is linked with consanguinity, however as trio-exome testing was not reported to have been performed in the previous 17 reported cases, no genetic links have been found. This case is the first to report a homozygous pathogenic CEP290 variant in GS.
- The prognosis is poor in GS, however, some literature suggests survival into and beyond the neonatal period can occur. Antenatal and postnatal MDT input is vital to manage parental expectations.
- If an infant with suspected GS survives beyond a few hours of life, early MDT input should be sought in order to assess what would be in the infant’s best interests regarding long-term manage-ment. The infant should be continually re-evaluated along the way.
Parental consent
The authors obtained written consent from the child’s parents for publication of the case history and images.
Or read this article in our
Tablet/iPad edition
- Goldston syndrome (GS) currently has no known genetic associations, but is thought to be a variant of Meckel-Gruber syndrome, the most severe of the CEP290-related ciliopathies.
- We present the first case of GS associated with a CEP290 variant.
- The prognosis is often poor for these patients and a multidisciplinary approach, including palliative care input, should be established postnatally.
Also published in Infant: