Simultaneous epidermolysis bullosa, pyloric atresia and imperforate anus in a newborn
The coexistence of congenital pyloric atresia (PA) and epidermolysis bullosa (EB) is rare but has been previously described. Here, we present the case of a newborn infant with EB-PA and an imperforate anus (IA). To our knowledge, the combination of EB-PA-IA has not been previously reported in the literature. We discuss the diagnosis and therapeutic measures in this unusual case.
*Fatemeh-Sadat Tabatabaei1
Pediatrician
*Seyyed-Mohsen Hosseininejad2,3
Resident of Orthopedics
Sahab-Sadat Tabatabaei3
Resident of Internal Medicine
Assistant Professor of Neonatology
ealaee@yahoo.com
1Rafsanjan University of Medical Sciences, Rafsanjan, Iran
2Neonatal and Children’s Health Research Center, Golestan University of Medical Sciences, Gorgan, Iran
3Shahid Beheshti University of Medical Sciences, Tehran, Iran
4Taleghani Children’s Hospital, Gorgan, Iran
*Joint first authors
The case
A 24-hour-old male infant presented to Taleghani Pediatric Hospital in Gorgan, Northern Iran, with non-bilious vomiting, failure of meconium defaecation and abdominal distension.
He was born at term (38 weeks’ gestation) weighing 3,400g by vaginal delivery to a 31-year-old gravida 5 female. The pregnancy and delivery were uncomp-licated with no history of polyhydramnios. Antenatal karyotyping was not carried out. His parents were non-consanguineous Turkmen with no family history of bullous disease or congenital anomalies.
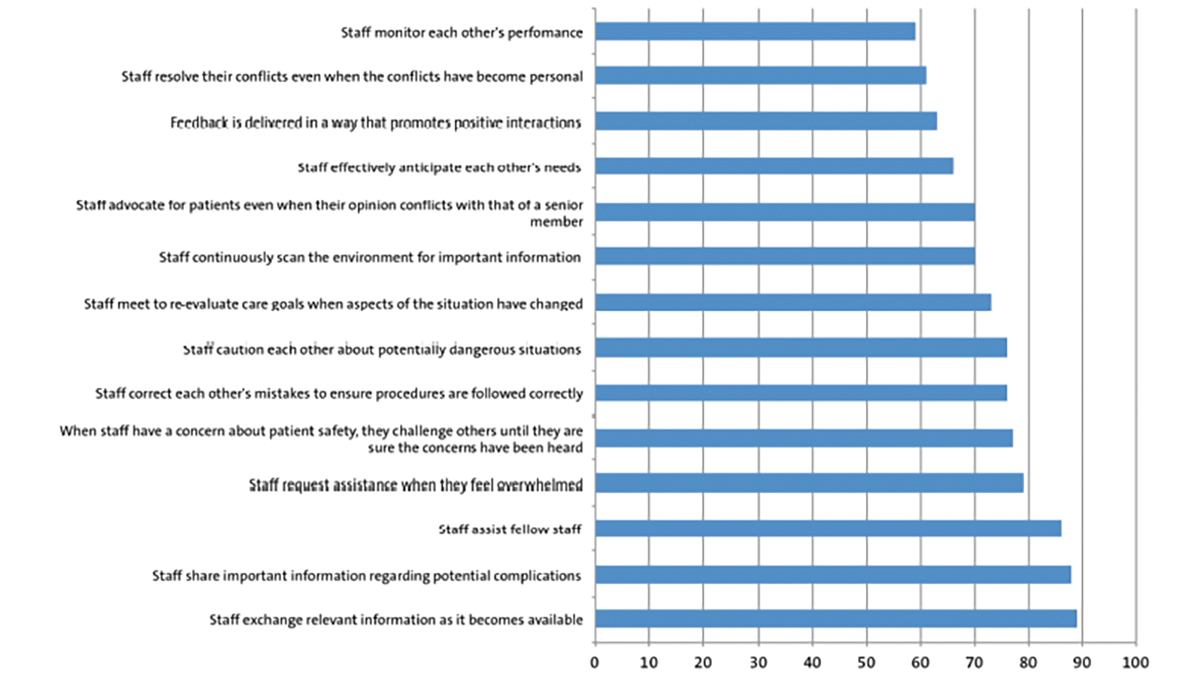
At birth his Apgar scores were 8 and 9 at one and five minutes, respectively. A physical examination revealed well-defined erythematous areas of peeled skin over the left forearm, left lower foot and scalp (FIGURE 1), and a membranous layer covering the anus, indicative of IA (FIGURE 2). Some very small blistering lesions were noted in the oral mucosa. A systemic physical examination showed no other abnormality except for a wide sagittal skull suture but with a normal head circumference of 34cm.
It was noted that the baby could not tolerate feeding. After initiating oral supplements through an orogastric tube, non-bilious, straw-coloured, gastric fluid was evident. Following this, the patient developed recurrent vomiting, he did not defaecate and his abdomen became distended.
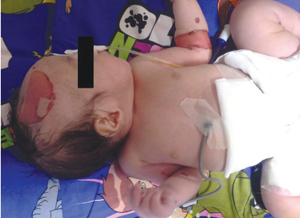
FIGURE 1 Well-defined erythematous areas of peeled skin indicative of EB.
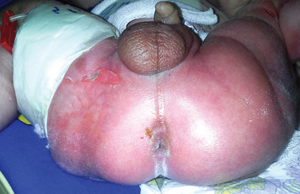
FIGURE 2 Imperforate anus: a membranous layer covering the anus.
Investigations
Laboratory blood tests were unremarkable. Abdominal X-rays illustrated gastric dilation with absent distal bowel gas and the duodenum was free of any air (FIGURE 3). Upper gastrointestinal barium meals showed a thickened and distended stomach (FIGURE 4); contrast failed to pass the pylorus to reach the duodenum. A distal colostogram depicted a dead-end lumen and no fistula of the rectum to the urinary tract or genitalia (FIGURE 5). Ultrasound of the abdomen did not reveal any renal or other organ abnormalities.
Histopathology of the skin biopsies obtained on the first and second days were compatible with a diagnosis of EB, ie dermal-epidermal cleavage and cell-poor subepidermal blisters.
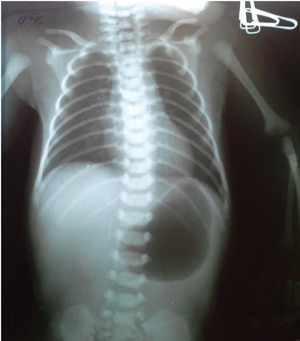
FIGURE 3 An X-ray showing gastric dilation with absent distal bowel gas.
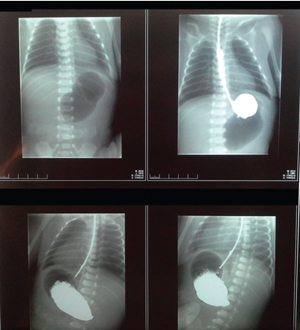
FIGURE 4 The contrast agent failed to pass the pylorus and did not reach the duodenum.
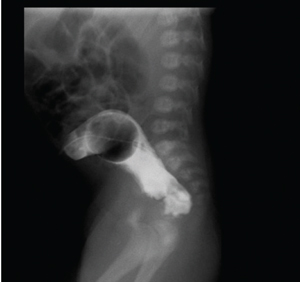
FIGURE 5 A distal colostogram depicting a dead-end lumen and no rectal fistula.
Treatment
Parenteral nutrition with amino acids, dextrose, lipid emulsion, nutritional supplements and electrolytes was provided. Following stabilising measures, a lapar-otomy was performed on day 3, which confirmed the existence of atresia of the pylorus, pouch colon and imperforate anus. The child underwent surgical procedures to correct the PA and IA – a gastrojejunostomy, direct percutaneous jejunostomy, a colostomy and a mucus fistula.
Skin care included lancing of blisters using a sterile needle, regular skin cleanings, dressing with topical antiseptics and antibiotics (ampicillin, amikacin and metronidazole). The dressings on broken skin were changed two to three times a week. All tapes on the skin were removed by waterlogging and liquid paraffin.
Postoperatively, the baby appeared more comfortable; his symptoms gradually subsided and he was discharged in a generally stable condition. At the one-year follow-up appointment his condition
was stable.
Discussion
The association of congenital EB-PA is very rare and, with few cases found in the literature, the exact prevalence remains unknown. Lestringant et al1 analysed 42 cases of EB-PA reported up until 1992. Worldwide, over 100 cases of EB-PA have been reported,2-6 however association of PA, EB and IA has not been reported to date as far as we are aware.
EB-PA
The clinical features of EB may be apparent at birth or may appear later. The skin may show localised or extensive areas of peeling or blistering with little or no trauma. Oral and mucous membrane involvement is common, as in the case described here. Congenital absence of skin (aplasia cutis congenita) is present in approximately 20% of cases7 but this was not seen in our case. Extensive EB lesions can have a fatal outcome in the neonatal period due to sepsis and fluid loss.8
PA is usually suspected when neonates develop recurrent non-bilious vomiting and abdominal distension; the distention is even more severe if IA coexists. Additional features of EB-PA may include fusion of the skin between the fingers and toes, nail dystrophy, scarring alopecia, contractures and dilated cardiomyopathy. Extra-cutaneous manifestations include involvement of genitourinary, respiratory and gastrointestinal tracts, in particular, genitourinary malformations such as dysplastic/multicystic kidneys, hydronephrosis/hydroureter, ureterocoele, duplicated renal collecting systems and an absent bladder,9 although such abnormalities were not detected in this case. Regular follow-up is needed in monitoring urinary symptoms because ureterovesical obstruction can occur after the neonatal period. Atresia involving multiple regions of the gastrointestinal tract may occur simultaneously.10,11 Polyhydramnios, secondary to PA, is usually evident by the third trimester in pregnancies with an affected fetus. In this case polyhydramnios was not reported.
The course of EB-PA is usually severe and often fatal in the neonatal period; coexistence of IA may lead to faster deterioration of the condition.
IA
The signs of IA include: no anal opening; an anal opening in the wrong place; no stool in the first 24-48 hours of life; stool passing through the wrong place, such as the urethra, vagina, scrotum, or the base of the penis; swollen abdomen; and an abnormal connection (fistula) between the baby’s rectum and reproductive system or urinary tract.7,12,13 The latter sign was not seen in our case.
Inheritance
EB-PA is inherited as an autosomal recessive disorder and is associated with mutations in three genes:11,14-16
- ITGB4 (integrin-β-4) – accounts for 80% of EB-PA patients
- ITGA6 (integrin-a-6) – 5% of patients
- PLEC1 (plectin) – 15% of patients.
In most cases the cause of an anorectal malformation is not known; the genetic basis of these anomalies is very complex because of their anatomical variability. In 8% of patients genetic factors are clearly associated and the gene HLXB9 may be involved.1,12,16
Diagnosis
EB can be diagnosed by skin biopsy using transmission electron microscopy and/or immunofluorescent antibody/antigen studies. Molecular genetic testing is available but not necessary to confirm the diagnosis. PA is diagnosed by the manifestation of a single gastric gas bubble on plain abdominal X-ray representing pyloric obstruction.17 IA should be picked up in a physical examination shortly after birth. A number of diagnostic investigations including X-rays, ultrasound and MRI will help to further evaluate the problem and determine whether other abnormalities such as spinal cord disorders are present.13
For at-risk pregnancies, prenatal diagnosis of EB-PA can be established by examining DNA or by using monoclonal antibodies.18,19 With the routine use of prenatal ultrasound scan, polyhydramnios and fetal gastric dilation may be detected long before the time of delivery.
If a prenatal diagnosis is established, a caesarean section should be planned to reduce trauma during delivery.
Genetic counselling is essential for families with a strong history of EB and/or PA and/or IA. Molecular genetic testing of the parents can establish heterozygous carrier status. Preimplantation genetic diagnosis for pregnancies at increased risk of EB-PA is possible once the gene mutations have been identified.
Management
Treatment of such disorders is mostly symptomatic and supportive. Surgical intervention is needed to correct PA and IA. Parents need education and training to better handle their infant. Skin care is of vital importance; protection of skin from poorly fitting clothing and use of atraumatic sterile dressings may avoid further skin peeling. Supportive measures improve outcomes, including antibiotics and antiseptics to prevent wound infections, fluid and electrolyte balance and nutritional support. Prevention of secondary complications such as growth delay, anaemia, zinc deficiency, osteopenia/osteoporosis and scarring of skin and mucosal surfaces should be considered. Psychological support for parents and caregivers is essential.
Prognosis
In most cases, the course of EB-PA is relentless and usually fatal despite surgical correction of PA. The prognosis is poor due to prematurity (although not in this case), extensive skin blistering with fluid and electrolyte imbalance, respiratory distress syndrome, malnutrition, sepsis, persistent vomiting and diarrhoea, and possible associated genitourinary disease.
In those cases who survive, the condition may improve with time. Some affected individuals may have little or no blistering later in life. Conversely, many affected individuals living past infancy experience blistering and formation of granulation tissue around the mouth, nose, fingers and toes, and infrequently in the trachea yielding stridor.
Conclusion
PA should be considered in neonates born with vesico-bullous lesions on the skin. Genetic counselling should be provided to families who are at risk for developing EB-PA. Adequate and timely medical and psychosocial care must be provided to reduce morbidity and improve the prognosis and quality of life. The course of EB-PA is usually severe and often fatal in the neonatal period; it is uncertain how the coexistence of IA may impact but it is possible that it may lead to faster deterioration of the condition.
Parental consent
The authors obtained written informed consent from the child’s parents for publication of the case history and images.
Or read this article in our
Tablet/iPad edition
- The case of a newborn baby with EB, PA and IA is described. This association has not been reported elsewhere.
- The course of EB-PA is usually severe; coexistence of IA may lead to faster deterioration of the condition.
Also published in Infant: