Enteral tube feeding in infants
Oral feeding (breast or bottle) is the preferred route for feeding babies, however there are a myriad of conditions that may necessitate enteral (tube) feeding. The more common conditions include prematurity, severe gastro-oesophageal reflux, failure to thrive, neurologic and neuromuscular impairment and anatomical congenital malformations. In recent years the range and ease of enteral access procedures have greatly improved; this article will review new techniques, equipment and accessories.
Katherine BurnandMBBS, FRCS
Locum Paediatric Surgery Consultant,
St George’s University Hospitals NHS Foundation Trust, London
Joe Curry
MBBS, FRCS(Eng), FRCS(Paed Surg),
PGDip Med Ed
Consultant Neonatal and Paediatric Surgeon
Great Ormond Street Children’s Hospital NHS Foundation Trust, London
joe.curry@gosh.nhs.uk
In order for enteral feeding to be successful the child must have a functioning gastrointestinal tract, otherwise parenteral nutrition may be necessary. Various artificial tube feeding options are available with nasogastric tube (NGT) feeding by far the most common.
There has been a global transition to specific enteral device connectors (no longer Luer lock) since the International Standards Organisation1 published a series of standards concerning small bore connectors in a range of medical devices to reduce the risk of cross connections of devices intended for different clinical applications. The EnFit connector is to be used for all enteral device connectors (FIGURE 1).2

FIGURE 1 An NGT with EnFit connector.
Nasogastric tubes
NGTs are quick and easy to insert and establish feeds through. They can also be used as drainage devices and for delivering medication. All healthcare professionals and healthcare assistants can insert NGTs once they have been trained and deemed competent. Trained parents and carers can also insert NGTs, which is particularly useful in the community. This is only possible after a full multiprofessional- supported risk assessment is performed and the local community team is informed prior to discharge of the child.
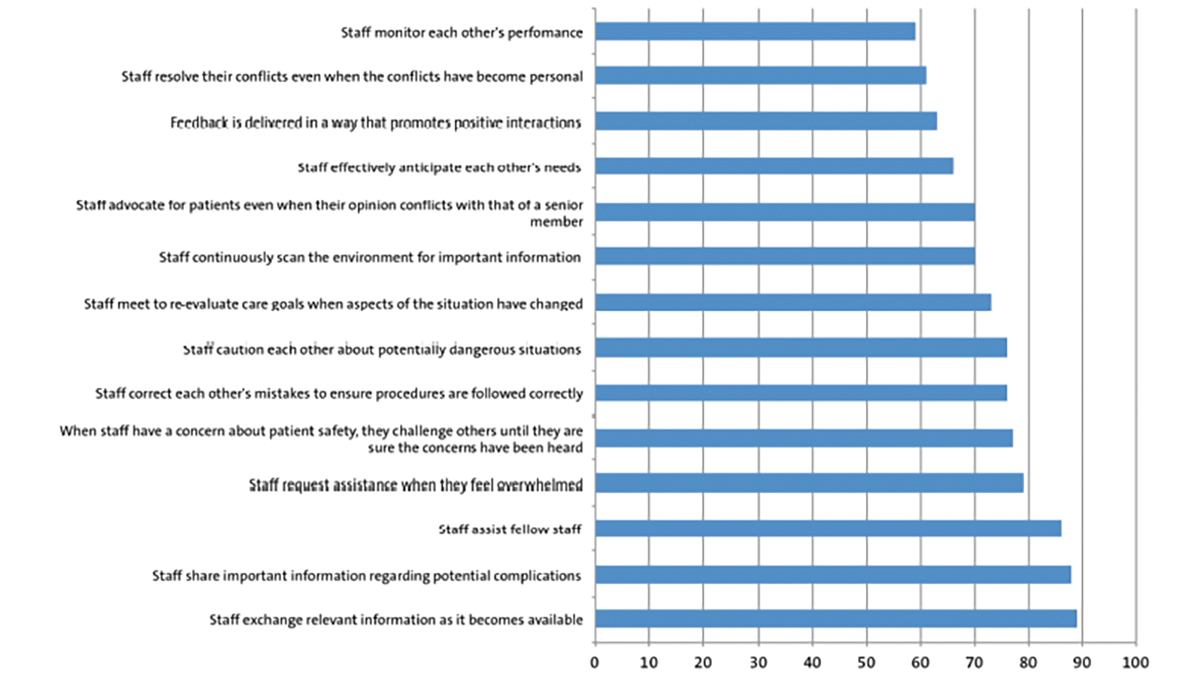
To work out how far the NGT should be inserted, the distance from the tip of the nose of the baby to the ear lobe plus the distance from the ear lobe to midpoint between the xiphoid process and umbilicus (FIGURE 2) are measured. Typically the range in sizes for the paediatric age group is 6-10 French. Wide bore NGTs are used typically for drainage and are stiffer than softer longer-term feeding NGTs. There are, however, a number of drawbacks to NGTs. Infants are likely to pull them out making regular re-insertion necessary. Securely taping the tube to the face is imperative to minimise the distress of re-insertion. Long-term use can result in damage to the skin on the face and in particular the nostril.
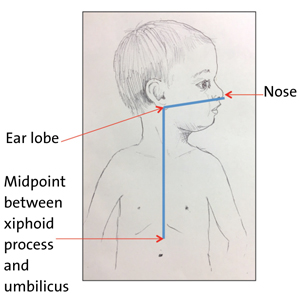
FIGURE 2 To ensure the NGT is inserted to the correct distance remember the acronym NEMU – nose, ear, mid-umbilicus (midpoint between the xiphoid process and the umbilicus).
The most serious complications related to NGTs have arisen from misplaced tubes, even resulting in patient deaths. The ‘gastric’ position of the tube must always be confirmed after insertion, before any feed/fluid or medication is instilled down the tube, at the change of feed if the child is receiving continuous feeds, or if there is any evidence of displacement. Tube position is checked with either pH testing of aspirate (safe pH = 1-5.5), or by X-ray if no aspirate can be obtained (after secondary manoeuvres) or the aspirate remains outside of the safe range after manipulation. Malpositioned tubes were first recognised as a patient safety issue by the National Patient Safety Agency (NPSA) in 20053 and further alerts have been issued by NPSA and NHS England.4-6 Introducing fluids, feeds or medications into either the pleura or respiratory tract via a misplaced tube is considered to be wholly preventable and therefore is now a ‘never event’.7 Misinterpretation of X-rays by medical staff is the most common error type (FIGURES 3 and 4).
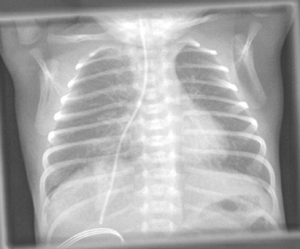
FIGURE 3 An example of a misplaced NGT down the right main bronchus.
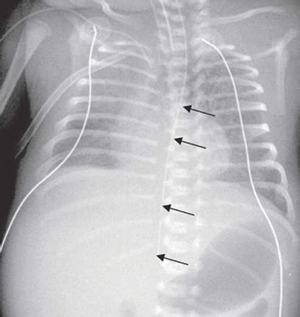
FIGURE 4 An example of an NGT taking an abnormal route, likely false passage due to iatrogenic injury to the oesophagus. The direction of the NGT is too straight and does not follow the normal gastro-oesophageal junction angle into the stomach.
Orogastric tubes
Orogastric tubes (OGTs) are similar to NGTs except they pass down into the stomach via the mouth. Indications for these include choanal atresia, use of nasal continuous positive airway pressure (nCPAP) ventilation prongs (in certain instances), suspected base of skull fracture or craniofacial anomaly.
Nasojejunal tubes
In some infants, feeding gastrically may not be suitable or safe in conditions such as severe gastro-oesophageal reflux (GOR, especially when complicated by acute life-threatening events or aspiration pneumonia), delayed gastric emptying or persistent vomiting.
Jejunal feeding is associated with a higher incidence of gastrointestinal infection and a sterile technique is necessary when handling the feeding sets. Continuous feeding is required as the jejunum is a tube and does not possess the reservoir function of the stomach. Bolus feeding into the jejunum would be painful and possibly result in ‘dumping syndrome’ due to too large an osmolar load being delivered straight into the small bowel resulting in diarrhoea and hypoglycaemia.8
Nasojejunal tubes (NJTs) are harder to insert and often need fluoroscopy or interventional radiology to aid placement. Similarly to NGTs, they can get displaced but will require hospital admission or even hospital transfer to reinsert. They can also block easily, which may necessitate declogging with a cocktail of enzymes, rewiring or even replacement. In young infants there is a non-image guided method for inserting them, which sometimes works by gastric peristalsis pushing the jejunal portion through the pylorus (FIGURE 5). An X-ray is still required to confirm the final position.
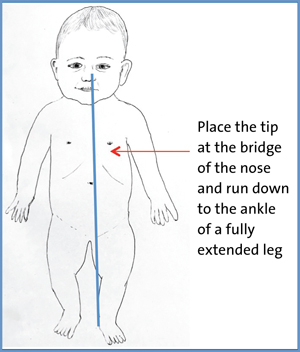
FIGURE 5 The method for measuring the length of NJT needed for insertion into the stomach. Measure from the bridge of the nose and run down to the ankle of the fully extended leg.
Gastrostomy
A gastrostomy is an artificial connection between the abdominal wall and the stomach maintained by an artificial device. It can be placed through operative intervention either laparoscopically or at open laparotomy, endoscopically or radiologically using real-time biplanar fluoroscopy. The percutaneous endoscopic technique (PEG) was first performed in a four-month-old baby by Gauderer et al in 1979.9
The indications for a gastrostomy will mirror closely those for an NGT. The main issue is the likely length of time that full or partial gastric feeding might be required. A gastrostomy should be considered if the time of gastric feeding is likely to exceed three months. Neurologic causes include cerebral palsy or unsafe swallow; psychological diagnoses such as food aversion or avoidance; or conditions resulting in failure to thrive, for example renal impairment, cystic fibrosis or metabolic syndromes. There are some instances in which NGT feeding is impossible or unsafe where there is a mechanical obstruction to the oesophagus such as oesophageal atresia without a fistula, vascular malformation, caustic stricture, epidermolysis bullosa or a tumour.
Primary PEGs are the most commonly inserted gastrostomy tube (FIGURE 6) using the Seldinger technique. They have been demonstrated to be safe even in the <5kg population.10 The stomach is insufflated with air through the endoscope then a clear indentation in the stomach of a finger pressing on the anterior abdominal wall should be observed before passing a needle through into the stomach followed by a guide-wire, which is then snared from the mouth. The guide-wire is removed through the mouth and attached to the gastrostomy tube, which is subsequently pulled back out through the abdominal wall leaving the flange (of the gastrostomy) flush with the stomach wall internally. The gastrostomy is then secured with a clamp close to the skin to keep the stomach up against the body wall.
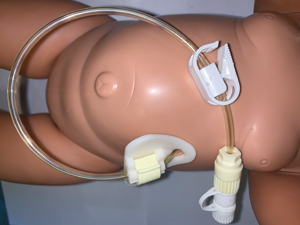
FIGURE 6 A percutaneous endoscopic technique set on a manikin.
The primary PEG technique may be unsuitable in patients who have undergone previous abdominal surgery, have significant hepatosplenomegaly, are on peritoneal dialysis or where it is not possible to obtain a clear indentation in the stomach with a finger. Laparoscopy can assist both the PEG and gastrostomy tube techniques and help identify the exact position of the gastrostomy placement on the stomach and minimise damage to other structures.
Open Stamm gastrostomy can also be performed safely using a small upper midline incision and placing a gastrostomy tube into the stomach via a separate incision, securing it in place with two purse string sutures and sutures to hold the stomach up against the abdominal wall. Primary balloon button gastrostomies can also be placed with the aid of T-fasteners to secure them to the abdominal wall. Radiologically inserted gastrostomies are also performed in certain paediatric centres with ready access to interventional radiology.
Once gastric function has returned feeds should be recommenced on a regime appropriate for the child. The infant should only be discharged once the parents are trained and able to deliver the feeding regime. Infants with a gastrostomy should have a nurse specialist as a point of contact. PEG tubes typically remain in situ for a minimum of six months and are usually changed within 18-24 months to either a button balloon gastrostomy or another PEG. Gastrostomy tubes inserted into a track that has been sutured can be changed after six weeks to button gastrostomy once the track has matured. Balloon gastrostomies should be changed every 3-6 months with the water content within the balloon checked weekly.
Minor complications such as granuloma formation, leak or infection are common with gastrostomies and most can be managed with minimal interventions such as bolstering techniques, silver nitrate, topical agents (steroids, antibiotics) or changing the tube (different length or gauge).
The tube can also block, break, displace or fall out. If the track is mature it is imperative that the tube is replaced swiftly either with another gastrostomy tube or, if one is not available, a similar gauge NGT through the gastrostomy. If the tube gets dislodged within the first six weeks of the initial insertion, blind re-insertion should be avoided as there is a risk of creating a false passage.
Buried bumper syndrome is another complication where the flange or balloon becomes embedded within the gastric wall making it potentially hard to instill fluids into the stomach. There is often pain and swelling at the exit site of the gastrostomy. This is particularly a problem with PEGs when they are left in place for two years or more. Regularly twisting and pushing in and out of the PEG minimises the chance of this happening.11
Gastrostomies can be left to close spontaneously if the child is no longer using them for at least three months and is thriving. Occasionally they require formal surgical closure.
Jejunal tubes
When it is not possible to establish gastric feeds it may be necessary to introduce post-pyloric feeding as a more permanent option, such as in severe GOR especially if the patient has already failed an anti-reflux procedure or in certain cases where the patient has no stomach (eg oesophageal replacement with stomach). As mentioned previously, jejunal feeding always requires continuous feeding as the jejunum is not a reservoir and cannot cope with large osmolar loads. Both trans-gastric and surgical jejunostomies come with their own array of benefits and problems.12
Gastrojejunal tubes
Trans-gastric jejunal feeding tubes (GJTs) have the advantage that they can be inserted through a pre-existing gastrostomy. There are typically two types: the PEG-J and the balloon button device. The balloon GJT does not typically require a general anaesthetic to be sited. The GJT has gastric and jejunal ports and therefore the stomach can still be accessed through the same device. The child will only have one ostomy on their abdominal wall. GJTs cannot typically be inserted in children <10kg. The disadvantages of the GJT are the reliance on hospitals for replacements and difficulty re-siting them out of hours, thereby necessitating urgent hospital admissions for intravenous fluids when they become displaced/blocked. They also require an interventional radiology service and each change will expose the patient to further radiation. Although it is less likely to get displaced, the PEG-J is at an increased risk of buried bumper as, unlike a standard PEG, rotating the device is not possible with the jejunal extension.
Surgical jejunostomy
There are different options for surgical jejunostomy when an infant is going to need long-term jejunal feeding and they cannot or do not tolerate a GJT.
Witzel tube
The Witzel tube is a form of jejunostomy where the bowel wall is imbricated over the tube creating a serosal tunnel. This has the advantage of no anastomoses and it can be performed laparoscopically, however the channel can be difficult to catheterise (requiring radiology) and it can be challenging in small infants to avoid narrowing the lumen of the bowel significantly, thereby creating a sub-acute mechanical obstruction. It is not amenable to a balloon jejunostomy due to further narrowing of the lumen. Correct positioning after tube changes should always be confirmed radiologically.
Roux-en-Y jejunostomy
This form of jejunostomy can accept a balloon jejunostomy at the first change and can be easily catheterised. This enables parents/carers to do these changes in the community. The downside is the jejunostomy requires an enteric anastomosis with potential for leak. There is a potential for volvulus of small bowel around the jejunostomy, which can be minimised by shortening the length of the Roux-en-Y limb.
Conclusion
In recent years the range and ease of enteral access procedures has greatly improved with the advent of new techniques, equipment and accessories. Enteral nutrition is not only more physiological than parenteral nutrition but it also avoids complications such as line sepsis, venous thrombosis and liver failure. Nonetheless tube assisted enteral feeding comes with its own array of complications that can be significantly reduced with appropriate training, knowledge and specialist gastrostomy nurses liaising between teams in the community and local hospital.
Or read this article in our
Tablet/iPad edition
- Enteral access procedures have greatly improved in recent years due to the availability of new techniques, equipment and accessories.
- Enteral feeding is associated with an array of complications but these can be reduced through appropriate training and specialist knowledge.